

Сохранение баланса при производстве стерильной продукции
При валидации процессов производства стерильных продуктов для демонстрации летальности методов стерилизации для микроорганизмов требуется использование биологических индикаторов. Индикаторные организмы существуют только для определения эффективности процесса стерилизации. В настоящее время повышенное внимание, уделяемое летальности биологических маркеров, приводит к использованию экстремальных условий при проведении стерилизации. Настойчивое предъявление повышенных требований к летальности при валидации стерильных процессов угрожает безопасности пациента. Проблема заключается не только в том, что при использовании подобной практики технологическое оборудование и сырье подвергаются воздействию повреждающих условий. Из-за подобных требований производители могут предпочесть использование асептического производства в случаях, когда финишная стерилизация является наиболее оптимальным и безопасным методом. Производители должны иметь свободу при выборе наиболее оптимального способа гарантировать безопасность процесса как при асептическом производстве, так и при финишной стерилизации или комбинации двух этих методов.
С середины 70-х годов прошлого столетия проведение валидации является обязательным для всех процессов, которые непосредственно влияют на микробиологическую безопасность материалов, используемых при производстве стерильных продуктов. Авторы обеспокоены тем, что в валидации в целом и особенно в области стерилизации строгость часто проявляется не там, где она требуется. В результате предъявляются чрезмерно завышенные требования на основании того, что «чем больше делается, тем выше безопасность». Например, вероятность наличия нестерильной частицы, составляющая 10-9, считается «лучше», чем 10-6, однако если исходить из соображений безопасности пациента, то чрезмерная строгость не имеет решающего значения и, следовательно, нецелесообразна с практической точки зрения. На практике неукоснительное соблюдение строгих требований исключительно ради строгости может привести к повышению риска для пациента, так как финишная стерилизация или другие способы снижения рисков в результате становятся менее доступными для производителя, что будет обосновано далее в данной статье.
Нормативные требования не совпадают с результатами достижений науки и техники. Учитывая, что оборудование, способы технологического контроля и технические возможности непрерывно совершенствуются, традиционные ожидания в отношении технологического контроля и валидации при этом не оправдываются. Использование традиционных методов при стерилизации и мониторинге параметров окружающей среды, таким образом, приводит к ошибкам.
В последние два десятилетия были разработаны правила и требования, которые опираются на ошибочную веру в то, что их внедрение будет способствовать повышению безопасности. В действительности же все, к чему они привели, – это снижение внимания к самому процессу, повышение стоимости производимых продуктов и усиление вероятности того, что абсолютно безопасный продукт может считаться небезопасным.
Сравнение подходов к стерилизации
С момента начала эпохи валидации в медицинской отрасли процесс стерилизации сосредоточился на повышении надежности процессов летальной стерилизации. Такой приоритет был обоснованным в 70-е годы прошлого столетия, когда сама по себе концепция валидации была в новинку. Однако после того как технические возможности процесса, его устойчивость и надежность были определены и валидированы, дальнейшие успехи не могут быть достигнуты путем повышения требований и внедрения более жестких регуляторных норм. Цель «большей уверенности» в процессах стерилизации преследуется без доказательств ее необходимости, что проявляется в излишних ограничениях, которые не способствуют повышению безопасности пациента.
Например, удлинились временные циклы процесса, увеличились минимальные требования к летальности и появились дополнительные (и иногда сомнительные) методы контроля в отношении практически всех аспектов процессов стерилизации. Эти изменения привели к необоснованному повышению летальности процесса, при котором не был получен ответ на вопрос: останутся ли стерилизуемые материалы пригодными для использования по назначению?
Все стерилизуемые продукты, за исключением нержавеющей стали и некоторых других материалов, подвергаются некоторым негативным воздействиям в ходе процесса стерилизации. Например, чрезмерный нагрев может отрицательно воздействовать на химические / физические свойства фармацевтических продуктов, уменьшая срок их годности и повышая распадаемость. Нагрев также может неблагоприятно воздействовать на стеклянные контейнеры, являясь причиной появления фрагментов стекла в растворе, а также разъедания и выделения повышенного количества частиц. Экстремальные условия могут приводить к изменениям физических свойств эластомерных материалов (например, пробок, уплотнителей, шлангов, фильтров) с потенциальными изменениями показаний дюрометра. Избыточное облучение является причиной существенных физических и химических изменений, происходящих почти во всех материалах. Длительная газовая, жидкостная или паровая стерилизация может обусловить коррозию, избыточную адсорбцию и химические реакции в материалах и оборудовании. Все более ограниченный размер пор фильтра снижает производительность, повышает рабочее давление и содержание экстрагируемых / выщелачиваемых веществ в жидкостях.
Можно действительно повысить безопасность процесса, но требуемая летальность бесполезна за определенной чертой. В пищевой отрасли применяется именно такой подход к стерилизации, что делает его приоритетным для сохранения органолептических свойств пищевых продуктов. В медицинской промышленности, однако, эту проблему игнорируют, и воздействие жестких условий процесса на материалы редко учитывают – только при финишной стерилизации влажным теплом или облучением.
Избыточная стерилизация может обеспечивать значительную логарифмическую редукцию спор высокорезистентных биологических индикаторных микроорганизмов, но при этом создает две проблемы:
- • Негативное воздействие на качество продукта, срок годности и другие его свойства вследствие того, что использование слишком жесткого процесса приводит к повреждению материалов.
- • Повышение риска для пациентов в случаях, когда производитель выбирает асептический процесс вместо финишной стерилизации, являющейся более безопасной.
Правильный подход к стерилизации должен гарантировать равновесие между необходимостью обеспечивать летальность для микроорганизмов и сохранением параметров качества материала. Достижение этого баланса требует более глубокого знания процесса стерилизации, точнее говоря, ее воздействия как на микроорганизмы, так и на стерилизуемые материалы.
Некоторое время назад Фармакопейная конвенция США приняла меры для нейтрализации негативных последствий чрезмерной обработки во время стерилизации путем концентрации большего внимания на предварительной стерилизации бионагрузки. Рутинный процесс стерилизации должен быть направлен на уничтожение бионагрузки до вероятности не более одной нестерильной частицы на 1 000 000. Подлинность, количество и резистентность биологических индикаторов менее важны, потому что индикатор не определяет процесс стерилизации, а, скорее, измеряет его. Процессы стерилизации должны быть не более летальны, чем необходимо для того, чтобы воспроизводимо уничтожить бионагрузку до безопасного уровня, в то же время максимально сохраняя наиболее значимые параметры качества. Все же микроорганизм можно убить только единожды. Мы можем говорить об избыточном уничтожении, но на практике можно ли еще раз убить микроорганизм, который уже мертв? Использование условий «наихудшего случая» в дополнение к уже существующим условиям «наихудшего случая» не приносит пользу никому. Любой процесс стерилизации, при котором время стерилизации (величина F0 ) превышает 10 – 12 мин, является чрезмерным. Он разрабатывался с учетом повышенного внимания к факторам (зачастую это реальные или мнимые регуляторные ожидания), которые не связаны с безопасностью пациента. Некоторые практикующие специалисты и представители регуляторных органов выдвигают ненаучные нормативные требования, не понимая, как действуют микробиологические принципы при стерилизации и как влияют на безопасность пациента или, возможно, они не учитывают в полной мере потенциальные негативные последствия чрезмерной обработки.
Существует возможность проведения безопасной стерилизации материалов и продуктов с использованием методов, которые находятся далеко за пределами сегодняшних требований к летальности. Особое значение имеет то, что отрасль остается открытой к научному и техническому прогрессу и таким его направлениям, как снижение температуры обработки и гарантирование наилучшего эффекта для пациента.
Асептическое производство: гарантированно безопасное, но не стерильное
Асептический процесс, который является безопасным благодаря исключению наличия микроорганизмов в продуктах, в значительной степени опирается на возможность стерилизовать и депирогенизировать материалы, из которых состоит стерильная дозированная форма. После стерилизации в тщательно контролируемых условиях используются уже стерильные предметы. Внедрение таких же, как в других отраслях, новых технологий в асептическое производство и улучшение в течение последних 20 – 30 лет функциональных показателей, таких как автоматизация и исключение участия персонала в процессе, будет, несомненно, продолжаться.
И несмотря на то, что усовершенствованные конструкции встречаются все чаще, современное асептическое производство осуществляется с использованием различных подходов – начиная от полностью ручных и заканчивая высокоавтоматизированными. За некоторыми, сравнительно немногими, исключениями всегда стабильно стерильные продукты производились с помощью систем асептического производства. Вопросы в отношении приемлемости асептического производства возникли на основании умозаключений, рожденных в результате наблюдений или работы с документацией. Контаминация продуктов, произведенных в асептических условиях, или, если более корректно, ощущение возможности контаминации, часто является результатом правильно высказанных, но научно не доказанных предположений со стороны экспертов регуляторных органов. Беспокойство вызывает то, что неоправданные ожидания могут привести к чрезмерно строгой интерпретации ограниченных, субъективных или аналитически неоднозначных данных, что в результате может вылиться в необоснованные утверждения о «недостаточном обеспечении стерильности».
В основании этих ожиданий лежит главная ошибка – что «асептический» означает «стерильный» или что окружающая среда должна быть стерильной для осуществления надлежащего асептического производства. В то время как в действительности стерильность в асептическом производстве была бы идеальным результатом, отрасли не хватает средств для получения доказательств того, что стерильность существует, поскольку невозможно объективно продемонстрировать полное отсутствие микроорганизмов. Ставить под сомнение целесообразность выполнения операций в асептических условиях, не являющихся «стерильными», означает игнорировать реальность.
Никакой объем наблюдений, моделирование процесса или контроль стерильности не могут гарантировать соблюдение стерильности в асептических условиях. Верным является и обратное утверждение – определение микробной контаминации проб из окружающей среды не означает, что загрязнение будет обнаружено в единицах продукции, произведенных в этой среде. Вероятность случайного загрязнения всегда существует, а отбор проб в асептических условиях как аналитически, так и статистически ограничен. Дальнейшая неопределенность возникает на основании всегда существующей вероятности того, что любой положительный результат анализа образца может быть ложным положительным результатом.
И хотя авторы уважают точку зрения тех, кто действует в интересах безопасности, на практике это может зайти слишком далеко. Целью асептического производства должна быть безопасность конечного потребителя, но безопасность и стерильность – это не одно и то же. Более высокий риск для пациентов может представлять непригодность продукта, а не предполагаемое, но в большинстве случаев недоказуемое «недостаточное обеспечение стерильности».
Асептическое производство стерильных продуктов – сложная задача, и для того, чтобы выдвигать концепцию Sterility by design (стерильность на этапе проектирования), необходимо учитывать большое количество факторов. Ниже перечислены следующие:
- • Мониторинг параметров окружающей среды, моделирование процесса и определение стерильности не являются контрольными мерами и только ограничивают значение параметров оцениваемого процесса и, в меньшей степени, технические возможности объекта оценки.
- • Успех асептического процесса заключается не в отсутствии микроорганизмов. Есть более важные и четкие факторы, необходимые для получения удовлетворительного результата.
- • Мониторинг окружающей среды не является достоверным способом определить безопасность продукта или уровень обеспечения стерильности и дальнейшее усиление интенсивности мониторинга не изменит этот факт.
- • Под успехом понимается возможность исключить из производственного процесса обслуживающего оператора и контролировать его вмешательство в процесс.
- • Асептическое производство может осуществляться более безопасно только благодаря усовершенствованию оборудования и технологического контроля. Обеспечение стерильности следует рассматривать как инженерные задачи, а не только как аналитическую микробиологическую проблему.
- • Вероятность загрязнения асептически произведенного в промышленном масштабе продукта в настоящее время оценивают как 1 на 10 000 единиц и ниже. Наличие более веских доказательств указывает на то, что это достаточно безопасно для здоровья людей.
В отрасли не должно существовать мнения, что асептическое производство, при условии использования современных достижений науки и техники в его осуществлении, является небезопасным или ограниченно безопасным. Возможно обоснованное опасение в отношении риска контаминации, если процесс включает проведение серии операций в открытых условиях или выполнение достаточно сложных процедур, либо если готовые дозированные формы производятся вручную без использования изоляторных технологий, таких как изоляторы. При осуществлении промышленных процессов асептического производства в соответствии с требованиями надлежащих производственных практик (cGMP) риск для пациента следует оценивать как низкий.
Оценка финишной стерилизации
Разница между асептическим производством и финишной стерилизацией всегда считалась существенной. Строгие догмы, которые определяют приемлемость процессов такой стерилизации, часто базируются на концепциях, которые в настоящее время являются иррационально консервативными и неуместными. Аналогичным образом на сегодняшний день очевидно, что не все технологии асептического производства одинаковы по своим возможностям. Отрасль находится на пороге нового понимания этих основных методов производства. Совершенствование технической оснащенности и более рациональное понимание микробиологии делают очевидными возможности применения более безопасных и экономичных способов производства стерильных продуктов.
Отрасль должна быть гибкой, чтобы разрабатывать процессы финишной стерилизации, имеющие больший смысл с точки зрения инженерии и микробиологической безопасности. Нормы или деревья принятия решений, которые ограничивают ученых отрасли при определении безопасных процессов, эффективных для пациента, являются примерами плохой регуляторной и нормотворческой практики. Мнение, что эффективная финишная стерилизация начинается при значениях времени стерилизации (величина F0) от 8 до 15 мин, является одновременно абсолютно неправильным и необоснованно ограничивающим. Наиболее вероятная причина такой строгости базируется на ложной вере в то, что резистентность биологических индикаторов (а, следовательно, их гибель) напрямую связана с безопасностью продукта; другими словами, нужно уничтожить как минимум миллион наиболее устойчивых спор, чтобы обеспечить вероятность наличия нестерильной частицы 10-6. Это недопонимание в сочетании с убежденностью в том, что ученые и инженеры, разрабатывающие технологии для отрасли, не в состоянии сделать правильный выбор как в отношении надежности процесса, так и безопасности пациента, является основой ошибочной логики, в соответствии с которой выдвигаются строгие и категоричные требования к стерилизации.
 |
Существующее мнение, что G. stearothermophilus, благодаря своей устойчивости к влажному теплу, необходим для демонстрации безопасности процесса финишной стерилизации, является достаточно очевидно неверным. Упорное применение G. stearothermophilus и длительных F0-циклов, которые являются следствием использования микроорганизма, повышают риск для пациента, потому что чрезмерные требования к минимальной летальности процесса стерилизации побуждают пользователей к выбору асептического производства. Это решение противоречит практически повсеместному предпочтению использования финишной стерилизации асептическому производству. Процесс финишной стерилизации, который, возможно, не в состоянии убить огромные количества непатогенных микроорганизмов G.stearothermophilus (маловероятная культура микрофлоры), но может стабильно уничтожать менее резистентные споры и все важные с медицинской точки зрения патогенные микроорганизмы, представляется в разумной мере безопасным. |
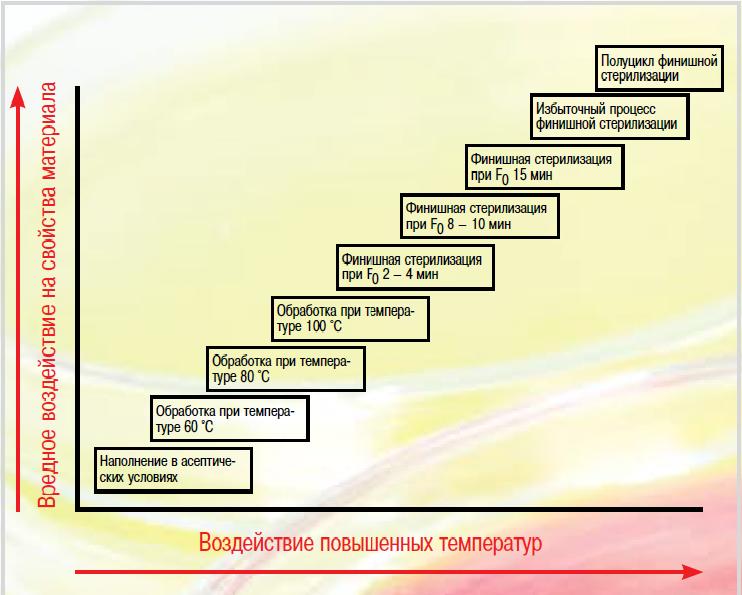
| Рис. 1. Модель процесса финишной стерилизации |
Таким образом, риск возникает из-за норм и положений, которые фактически препятствуют научному выбору условий стерилизации. В Японии был отмечен высокий уровень безопасности продукта при финишной стерилизации лекарственных препаратов при F0 2 мин и менее. Процесс стерилизации более не должен быть летальным для того, чтобы стабильно уничтожать любую существующую бионагрузку. На рис. 1 представлено новое решение в отношении выбора процесса финишной стерилизации.
Технологии асептического производства
В настоящее время используется много различных технологий асептического производства. При выпуске новых исследуемых лекарственных препаратов для проведения ранних стадий клинических исследований до сих пор используется большое количество ручных операций, но в промышленных процессах их не применяют, за исключением производства орфанных препаратов. Для всех ручных операций в фармацевтической промышленности следует применять
изоляторные технологии, но очевидно, что переход на изоляторы происходит быстрее при промышленном производстве.
До середины 80-х годов прошлого столетия, когда изоляторы только появились, асептическое наполнение осуществлялось в классифицируемых помещениях персоналом в специальной одежде, который выполнял операции по установке, а также вносил неизбежные корректирующие вмешательства. Изоляторы обеспечивают почти абсолютное отделение персонала от стерильных поверхностей и материалов и позволяет осуществлять автоматизированную деконтаминацию укупорочных материалов.
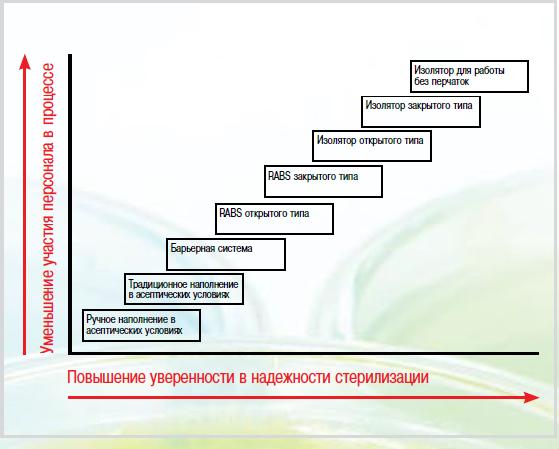 |
Внедряемые барьерные системы ограниченного доступа (RABS) приближаются по своим свойствам к исключительным возможностям изоляторов, в то же время сохраняя простоту обслуживания «чистых помещений» и исключая необходимость проведения деконтаминации газообразной перекисью водорода или аналогичным спороцидным веществом. Системы RABS отличаются более легкой конструкцией и привлекательным дизайном, почти не уступая изоляторам в возможностях, однако использование некоторых, менее совершенных конструкций RABS требует значительного вмешательства человека в изолированное пространство. Появляются новые технологии асептического производства – изоляторы для работы без перчаток и полностью закрытые системы, представляющие собой настоящие и будущие вершины производительности в асептических процессах. Многие из этих современных асептических технологий уже внедрены в мировой пищевой индустрии для фасовки в асептических условиях продуктов питания и молочной продукции. Можно прогнозировать их все более широкое применение в медицинской промышленности. Иерархическая схема асептических технологий изображена на рис. 2. |
| Рис. 2. Модель асептического производства. RABS – барьеры ограниченного доступа |
Определение стерильного производства
Производство стерильных продуктов не следует рассматривать как неизбежный выбор между финишной стерилизацией и наполнением в асептических условиях. Скорее стерильное производство продуктов необходимо понимать как целый ряд технологий, а их разработчики должны иметь свободу при выборе технологии или комбинации технологий, которая наилучшим образом обеспечивает как безопасность пациента, так и достижение коммерческих целей. Стандарты и регуляторные нормы, которые сужают выбор способов обработки без реального научного обоснования, только наносят вред производителю и потребителю. Авторы не видят оснований для разработки дерева принятия решений, которое подразумевает строгий учет температуры (летальной) при проведении финишной стерилизации.
Подавляющее большинство известных семейств бактерий, плесени, дрожжей и вирусов, являющихся причиной заболеваний человека, можно эффективно уничтожать при температуре гораздо ниже 100 °C. Список микроорганизмов, которые погибают при такой температуре, включает все грамположительные и грамотрицательные неспорообразующие бактерии, все вирусы, дрожжи и плесень. Таким образом, микроорганизмы, являющиеся возбудителями более 99% инфекционных заболеваний человека и животных, можно эффективно и успешно уничтожать при температурах значительно ниже тех, которые обычно используют для стерилизации. Любой разумный микробиолог или специалист по стерилизации сочтет это очевидным свидетельством того, что высокая безопасность процесса может быть обеспечена в значительно более щадящих, чем при F0 ≥ 8 мин, условиях.
Существуют спорообразующие организмы, являющиеся причиной заболеваний, и при разработке любого процесса стерилизации необходимо рассматривать риск для пациента, связанный с воздействием этих микроорганизмов. Наиболее хорошо известным спорообразующим патогенным микроорганизмом является Вacillus anthracis, возбудитель сибирской язвы. В общественном сознании сибирская язва ассоциируется с ее использованием террористами в качестве биологического оружия. Эти редкие патогенные споры являются наглядным примером наиболее устойчивого к воздействию тепла организма, образующего эндоспоры и представляющего интерес с медицинской точки зрения. Углубленное изучение термоустойчивости В.anthracis по сравнению с другими штаммами рода Вacillus было проведено группой авторов во главе с Монтвиллем (Montville).
Полученные результаты демонстрируют явное несоответствие требований, предъявляемых к минимальной допустимой температуре 121 °C и минимальному времени цикла 15 мин, и летальностью, необходимой для достижения объективной вероятности наличия нестерильной частицы 10-6 или ниже. Существуют очевидные доказательства того, что патогенные споры можно без труда и воспроизводимо уничтожать при температурах значительно ниже, чем те, которые представлены в дереве принятия решений PIC/S. Требования к длительности циклов, приведенные в дереве принятия решений, слишком консервативны и буквально исключают использование безопасных условий процесса. Более научным подходом было бы поощрение самостоятельного изучения условий стерилизации лекарственных препаратов без ограничений значениями температуры, считающимися эффективными в фармацевтической литературе.
С точки зрения авторов, величина летальности процесса, измеряемая в F0, должна восприниматься как один из вариантов выбора, а не как обязательное требование. В последние десятилетия, однако, существовала непоколебимая вера в обоснованность двух факторов, которые перевешивают в сторону летальности.
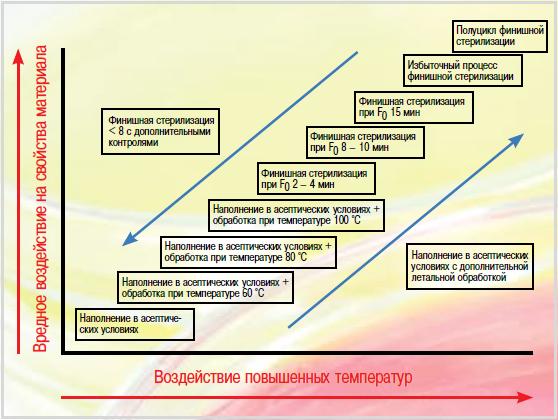 |
Один из них – температура 121,1 °C как обязательный параметр при стерилизации влажным теплом, а другой – обеспечение высокого уровня безопасности, который может быть достигнут только при уничтожении G. stearothermophilus. Данные в отношении резистентности различных патогенных организмов, включая В. anthracis, подчеркивают необоснованность этих убеждений. Температура 121,1 °C – это не более чем 250 ° по Фаренгейту (температура, эквивалентная давлению пара величиной 15 фунтов на квадратный дюйм), которую издавна использовали при автоклавировании консервированных продуктов. Принимая во внимание, что существенной летальности патогенных микроорганизмов, включая спорообразующие, можно достичь при температуре ниже 100 °C, не имеет абсолютно никакого смысла считать циклы стерилизации, проводимые при температурах между 121,1 и 100 °C (или, возможно, при более низких температурах), неэффективными с точки зрения летальности. Кроме того, учитывая эффективность температур в интервале между 70 и 100 °C для выявления неспорообразующих микроорганизмов, к которым относится подавляющее большинство возбудителей инфекционных заболеваний, циклы стерилизации при этих температурах в качестве дополнения к асептическому или даже тщательно контролирующему бионагрузку производству можно считать эффективными. На рис. 3 представлено несколько вариантов процесса, а также комбинации различных процессов. |
| Рис. 3. Модель стерильного производства, включающая асептическое производство (АР) и финишную стерилизацию (TS). Числовые значения на рисунке приведены в качестве иллюстрации и не являются определяющими; прогресс процесса следует понимать как непрерывный |
Сочетание асептического производства и финишной стерилизации
Проведение обработки в асептических условиях в качестве этапа, предшествующего финишной стерилизации, кардинально меняет концепцию. Такой подход обеспечивает тщательный контроль предстерилизационной бионагрузки, в результате чего может быть разработан последующий процесс стерилизации при более низких температурах (снижение общей летальности), что позволяет значительно расширить использование финишной стерилизации. Этот подход имеет явные преимущества с точки зрения безопасности:
- • Любой случайно попавший в контейнер в ходе асептического производства контаминант из бионагрузки можно легко уничтожить на этапе финишной стерилизации, значительно уменьшив объем или исключив необходимость проведения расширенного мониторинга параметров производственной среды.
- • Проблемы бионагрузки для финишной стерилизации по сути исключены, так как все флаконы наполнены в асептических условиях.
- • Если продукт был произведен с использованием только одного из процессов, то общеизвестные недостатки каждого из них (отсутствие высокой температуры, гарантирующей летальность, в асептическом производстве, разрушение продукта при финишной стерилизации) сохраняются.
Традиционные, взятые из данных дерева принятия решений целевые значения F0 и сочетания время-температура являются целями, определенными без достаточных оснований и предназначенными для упрощения осуществления процесса и его валидации, но они снижают гибкость, вследствие чего ограничивают возможности ученых, занимающихся вопросами стерилизации, в выборе более безопасных и легко контролируемых подходов. Надлежащие усилия должны быть сосредоточены на уничтожении микроорганизмов бионагрузки, а не на физических характеристиках; числовые значения по своей природе консервативны и не принимают в расчет воздействие процесса на продукт. Кроме того, показатель полноты удаления контаминантов должен быть использован для постоянной оценки трендов при мониторинге, а не для определения уровня тревог и действий, которое часто приводит к слишком строгому или неправильному истолкованию результатов.
Целью производства стерильных лекарственных средств всегда должно быть обеспечение пациентов безопасными и эффективными продуктами. От использования сочетания асептического производства и финишной стерилизации выиграют все. Пациент получит более безопасный и более стабильный продукт, максимально сохраняющий свои свойства, а у производителя будет меньше проблем в процессе производства. Регуляторные органы также выиграют, потому что безопасность пациента является их главной задачей, а потребитель будет оптимально защищен от риска.
Внедрение этих процессов не вызывает затруднений, так как оборудование для стерилизации можно модифицировать или использовать в существующем виде. Благодаря применению комбинированного процесса повышаются требования к валидации, но существенно уменьшаются проблемы, связанные с контролем контаминации в ходе асептического производства, и снижается необходимость в проведении обширного и дорогостоящего мониторинга. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) и Европейское агентство лекарственных средств (ЕМА) подписали документ, в котором сделан упор на использование финишной стерилизации, а также предлагается проявлять гибкость в процессе ее проведения.
Финишная стерилизация при температурах 70 – 80 °C может быть эффективной при условии проведения тщательно контролируемого асептического процесса. Обслуживание практически всех систем получения воды очищенной происходит при температурах выше 70 °C, и содержание микроорганизмов стабильно составляет около нуля колониеобразующих единиц. Однако асептическое производство не должно быть обязательным предварительным этапом перед проведением финишной стерилизации и не за всеми асептическими процессами должен или может следовать летальный процесс. Гибкость технологического процесса и регуляторных норм должны позволять ученым и инженерам выбирать оптимальные для производимого продукта условия обработки.
Еще одним преимуществом масштабного использования финишной стерилизации может стать более широкое применение выпуска по параметрам. Авторы соглашаются с мнением доктора Цугуо Сасаки (Dr. Tsuguo Sasaki), ранее являвшегося сотрудником Управления лекарственных средств и медицинских изделий Японии, который предположил, что выпуск по параметрам можно использовать в любом процессе финишной стерилизации, которая осуществляется при F0 > 2 мин. Учитывая статистическую и аналитическую лимитированность анализа на стерильность, снятие ограничений с использования выпуска по параметрам может иметь смысл.
Считается, что жесткие стандарты требуются потому, что в штате производителей нет специалистов, имеющих достаточную квалификацию или интеллектуальные способности для самостоятельного и правильного выбора способа обработки. Авторы объясняют это в равных долях паническим страхом и лицемерием. Такое утверждение является оскорблением для многих высококвалифицированных и опытных инженеров и микробиологов, которые работают в этой области. Возможно, подобное утверждение о всеобщей некомпетентности в качестве оправдания такого догматизма является удобным объяснением крайностей, существующих в регуляторных нормах. Какой бы ни была причина, тяжело найти какую-либо последовательность в выражении предпочтения финишной стерилизации при одновременном ограничении ее использования путем установления ненужных строгих стандартов и норм.
Еще одна странность заключается в том, что «проектное поле» между асептическим производством и финишной стерилизацией является, по существу, неисследованным. Существует, очевидно, некая мысль, учитывающая возможности асептического производства с последующей термической, радиационной или другой обработкой, которое позволит уничтожить подавляющее большинство патогенных микроорганизмов. Знание того, что лекарственный препарат можно безопасно обрабатывать без воздействия на его эффективность в течение 5 мин при температуре 70 °C, например, было бы важными сведениями, которые могут принести пользу пациенту / пользователю на всех стадиях жизненного цикла продукта, в том числе при производстве генериков, а также в аптечной практике. Игнорирование преимуществ такого подхода является недальновидным и противоречит всеобщей заботе о безопасности пациента, в которой заинтересованы и потребители, и производители, и представители регуляторных органов.
Использование асептического производства или производства, в ходе которого контролируется бионагрузка, в сочетании с финишной стерилизацией – не новая идея; в настоящее время существует множество примеров этих процессов. Учитывая преимущества такого сочетания, должно быть очевидно, что ограничения придуманы отраслью самой для себя. Наступило время для того, чтобы устранить деспотичные и ненужные разночтения в отношении асептического производства и финишной стерилизации и дать производителям право выбирать оптимальное решение для своих продуктов.
Подготовлено по материалам статьи, James Agalloco & James Akers, Achieving Balance in Sterile Product Manufacturing, Pharmaceutical Technology, 2015, Том 39, Выпуск 12
http://www.pharmtech.com/achievingbalance-sterile-product-manufacturing
Список литературы
- 1. FDA, 21 CFR 211, Proposed Good Manufacturing Principles for Large Volume Parenterals (Rockville, MD, June, 1976).
- 2. J. Agalloco, J. Akers, and R. Madsen, PDA J. Pharm. Sci. Tech. 52 (6) 346-350 (1998).
- 3. J. Akers and J. Agalloco, PDA J. Pharm. Sci. Tech. 55 (3) 176-184 (2001).
- 4. J. Agalloco, J. Akers, and R. Madsen, PDA J. Pharm. Sci. Tech. 63 (2) 89-102 (2009).
- 5. Her Majesty’s Stationary Office, Health Technical Memorandum 2010 (HTM- 2010) Part 3: Validation and Verification (London, 1994).
- 6. British Standards Institution, EN 285, Sterilization - Steam sterilizers - Large sterilizers; EN 285:2006+A2:2009 (London, June, 2006).
- 7. USP, USP General Chapter <1229>, “Sterilization of Compendia Articles” (US Pharmacopeial Convention, Rockvile, MD, 2013).
- 8. USP, USP In-process Draft <1229.5> “Biological Indicators,” Pharmacopeial Forum 41 (2) 2015.
- 9. J. Agalloco and J. Akers, Ed., Advanced Aseptic Processing Technology (InformaUSA, New York, 2010).
- 10. J. Akers and J. Agalloco, Pharm. Tech. 36 (5) s48-50 (2012).
- 11. EC, EudraLex Volume 4: Good manufacturing practice Guidelines, “Annex 1, Manufacture of Sterile Medicinal Products,” (Brussels, 2008).
- 12. PIC/S, Decision Trees for the Selection of Sterilization Methods (CPMP/QWP/155/96) (Geneva, 1999).
- 13
28.06.2016

